Properso Veesler


CRISTALLISATION EN SOLUTION
Stéphane VEESLER (E-mail)
Directeur de recherche au CNRS (section 05)Recherche au sein du département Sources et Sondes Ponctuelles
Permanents:
Thirou BACTIVELANE, Nadine CANDONI, Romain GROSSIER, Mehdi LAGAIZE.
Doctorants:
Celine Huart en cours
lien vers thèses soutenues
Post-Doc:
1.Approche Classique de la Cristallisation
La cristallisation en solution est contrôlée par un certain nombre de paramètres physico-chimiques tels le solvant/milieu de cristallisation, la sursaturation, la température, les additifs/impuretés, ainsi que l'hydrodynamique de la suspension. Les études que nous menons portent sur l'effet de ces paramètres sur les propriétés des solides cristallisés. L’aspect thermodynamique n’est pas laissé de coté puisque dans la majorité des cas, le produit final dépend fortement des conditions initiales, ce qui nécessite une très bonne connaissance du diagramme de phases.
Nous avons développé des méthodologies et technologies innovantes pour l'étude de la cristallisation de molécules organiques et biologiques avec pour contrainte de diminuer les quantités et les volumes manipulés de matériaux (adaptés aux faibles quantités usuellement rencontrées dans les phases de développement de nouvelles molécules organiques ou biologiques) . Nous avons développé un outil microfluidique polyvalent et facile à utiliser par les non-spécialistes pour le criblage des conditions de cristallisation, l'établissement rapide de diagrammes de solubilités et la mesure des cinétiques de nucléation des cristaux (volume nano-litre – premiers pas vers le confinement). Ces nano-cristallisoirs offrent des potentialités de contrôle intéressantes sur le plan des propriétés d’usage, par exemple obtention de cristaux calibrés, mais aussi d’un point de vue de la sécurité et de l’environnement. Ainsi, cette méthode permettra la conception de nanotechnologies innovantes pour la mise en forme et la stabilisation de cristaux, dans le but de fabriquer des matériaux à forte valeur ajoutée.
- Morphologie et faciès
Les additifs ou impuretés affectent tous les stades de la cristallisation. Ils peuvent conférer aux cristaux des morphologies et des faciès qui assureront ou non les propriétés d'usage désirées. La thèse de P. Taulelle (2003-2006) a porté sur l'utilisation des additifs de cristallisation et l'étude des mécanismes de croissance cristalline pour contrôler le faciès des cristaux d’une molécule d’intérêt pharmaceutique (collaboration avec SANOFI-AVENTIS) où l'approche expérimentale est développée en parallèle avec la modélisation moléculaire (fig.1). Les additifs peuvent aussi retarder l'apparition d'un polymorphe. Connaître et maîtriser l'impact de ces additifs représente donc un enjeu important.

Figure 1: Irbesartan forme A : (a) ) morphologie expérimentale (MEB), (b) morphologie prédite par les PBC, topographie par AFM faces latérales (b)et terminales (d).
- Mécanisme de croissance des cristaux aciculaires (article)
Dans ce cas la sélection d'additifs permettant de modifier le faciès des cristaux ne pourra être réalisée sans la compréhension au préalable des mécanismes de croissance des cristaux aciculaires. L'observation fine de nombreux cristaux aciculaires montre que les aiguilles sont généralement constituées d'agglomérats "orientés" d'aiguilles(fig.2). Il est donc intéressant pour décrire et interpréter ces résultats de comparer les mécanismes rencontrés lors de la croissance d'aiguilles et ceux proposés dans le cas des macles de croissance, où la formation se fait par "regular over et intergrowths". Deux mécanismes sont alors distingués :
Cas 1 : un mécanisme par syneusis, dans lequel les cristaux nucléent, croissent individuellement, puis s'agglomèrent suivant une direction particulière.
Cas 2 : un défaut de germination bidimensionnelle pour lequel une molécule s'incorpore de manière anormale au bord d'un gradin, et les molécules suivantes viennent s'assembler en position de macles suivant une orientation cristallographique particulière pour donner naissance à une macle. L'observation d'un angle rentrant sur le cristal de la figure 2d confirme cette hypothèse. De plus, aucune croissance des faces latérales n'a été observée avant l'apparition de la nucléation secondaire. Ceci implique que la formation de nouveaux cristaux nécessite moins d'énergie (c'est-à-dire une différence de potentiel, fournie par la sursaturation, moins importante) pour le système que la croissance couche par couche des faces latérales (par un mécanisme de croissance de type BCF ou par germination 2D). En ce qui concerne les faces terminales, les mesures montrent que la croissance nécessite à chaque fois une sursaturation critique avant de démarrer ce qui est cohérent avec un mécanisme de croissance par germination bidimensionnelle de type birth and spread.
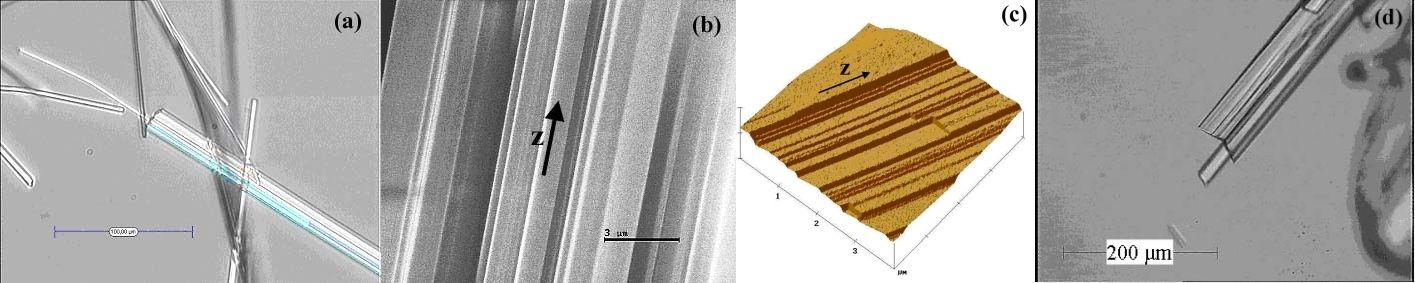
Figure 2: Cristaux organiques observés par: (a)et (d) microscopie optique, (b) MEB, et (c) AFM.
lien vers la thèse PASCAL TAULELLE
- polymorphisme (article)
La visualisation des phénomènes est réalisée par le système monopuit thermostaté par effet Peltier couplé à un microscope inversé (voir c'est comprendre!).
{kind=link}
- Démixtion et cristallisation (article)
Il se peut aussi qu’au cours d’une cristallisation par variation de température ou de composition, une démixtion (séparation de phase liquide-liquide) se produise; l’impact d’un tel phénomène sur la cristallisation peut aller jusqu'à inhiber la nucléation du solide désiré (fig.3a-c). Pour certaines conditions expérimentales, à fort refroidissement - pénétration dans la zone de décomposition spinodale, et après plusieurs heures, la nucléation primaire a lieu dans les gouttes. Puis par nucléation secondaire, croissance et agglomération dans les gouttes, on obtient des particules quasi-sphériques (fig. 3d-f).
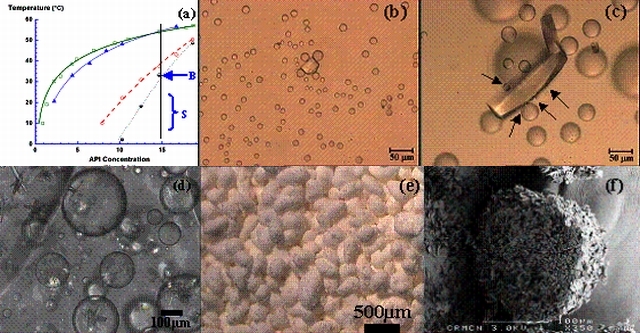
Figure 3 : (a) Diagramme de phases de C35H41Cl2N3O2 dans un mélange éthanol/eau (54.2/45.8% masse) les carrés creux = solubilité FI, les triangles pleins = solubilité FII, Les cercles creux = coexistence des phases liquides et les cercles noir et blanc = points de la spinode. (b) et (c) Observation par microscopie optique d’une cellule de cristallisation thermostatée ensemencée avec des cristaux de la phase I à la limite intérieure de la binode à 35°C, point B (fig.2a). Les flèches indiquent la nucléation hétérogène de gouttes à la surface des cristaux. (d) Cristallisation dans la zone S (fig. 2a), (e) particules obtenues à la fin de l'expérience et (f) Images MEB d'une coupe d'une particule.
Nous avons montré qu'une connaissance du diagramme de phases permet de transformer un inconvénient, une nucléation fainéante, en un avantage en développant une cristallisation dans les gouttes pour produire des particules sphériques. Les perspectives portent sur l'étude de la cristallisation en milieu confiné et la microfluidique.
lien vers la thèse LAURENT LAFFERRERE
- Criblage (article)
Nous travaillons aussi sur le développement de systèmes expérimentaux de cristallisation de petits volumes (15 à 100µL) adaptés aux faibles quantités usuellement rencontrées dans les phases de développement de nouvelles molécules organiques ou biologiques. Ce projet a donné lieu à la réalisation d'un montage expérimental original (voir service électronique) constitué d'une platine motorisée XY, équipé de cellules multi-puits (16 à 96 selon volume) maintenus à température constante par effet Peltier, d'un microscope optique, relié à un montage vidéo afin de procéder à l’acquisition séquentielle et au traitement des 96 (16) images pour étudier l'effet de la température et du milieu de cristallisation sur la nucléation, la croissance et les transitions de phases de cristaux en milieu stagnant (fig.4).

Figure 4 : Montage multi-puits
(article)- Comment mesurer une fréquence de nucléation ? ( microfluidique JCG2012), ( microfluidique CGD2011) ( microfluidique OPRD2012)
Il apparait clairement que l'étape de nucléation est clé dans le contrôle des propriétés des matériaux étudiés, qu'il s'agisse de macromolécules biologiques, de molécules organiques ou minérales. A ce jour deux théories sont utilisées pour discuter les résultats expérimentaux, la théorie classique (TCN), issue de la condensation d'une goutte à partir de la phase vapeur, ou la théorie en 2 étapes qui prend en compte le coté ordre de la transition de phase. C'est la raison pour laquelle un contrôle expérimental et une compréhension fine des mécanismes de nucléation ouvriront de nouvelles perspectives dans les recherches dans ce domaine des sciences des matériaux. Pour cela nous avons développé une approche en deux temps: (1) mesurer les fréquences de nucléation en solution en fonction de la sursaturation, de la température et du milieu et (2) agir sur la nucléation (voir "Approches non-classique"). Les expériences portent sur le lysozyme qui est utilisé comme substance modèle du fait de notre connaissance de son diagramme de phases et de notre expérience dans le domaine de la cristallisation des protéines.
L'obstacle majeur à la détermination de la fréquence de nucléation est la quantité importante de produit utilisé et le nombre d'expériences indispensables pour établir une statistique représentative de la nucléation (ceci est du à la stochasticité du phénomène de nucléation). Afin de réduire à la fois la quantité de produit et la durée de l'étude, on se sert de montages expérimentaux utilisant des volumes de l'ordre de la dizaine de µL en cellule de cristallisation et de la centaine de nL pour les gouttes générées par microfluidique pour lesquels les expériences sont menées en parallèle. En pratique il y'a deux méthodes de détermination de la fréquence de nucléation mesurent du temps d'induction ou détermination directe de l'état d'équilibre de la fréquence de nucléation homogène. La première méthode consiste à établir la sursaturation, puis à mesurer le temps qu'il faut pour pouvoir observer les cristaux, est de dissocier la première étape de nucléation de celle de la croissance par la méthode de la double pulsation thermique,donnant ainsi accès au contrôle de la nucléation. Au début d'une expérience, la solution est stockée à une température choisie afin d'éviter la nucléation des cristaux. Ensuite, la température est baissée jusqu'à une température TN à laquelle se produit la nucléation. Après un temps tN la température est élevée de la température de nucléation TN à la température de croissance TC (fig.5-1a et b). A TC, la sursaturation est suffisamment faible pour que la fréquence de nucléation soit pratiquement nulle, mais les cristaux déjà formés croissent jusqu'à devenir observables. Cela permet de séparer la nucléation de la croissance. Après l'étape de croissance, les cristaux nucléés pendant tN sont comptés. Après avoir tracé le nombre de cristaux nucléés en fonction de tN, la fréquence de nucléation est déterminée par la pente de la droite obtenue (fig.5-1c et d).
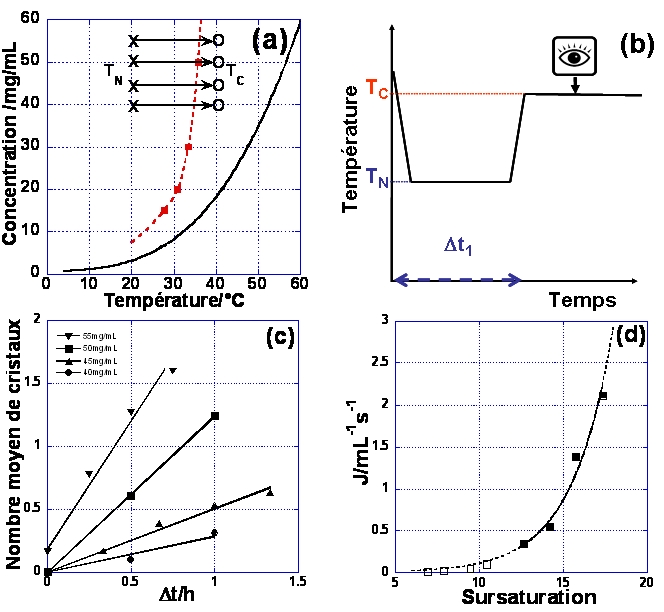
Figure 5-1 : fréquence de nucléation par double pulsation thermique (a) courbe de solubilité du lysozyme (0.7M NaCl - pH=4.5) et zone métastable
(X) et (O) indiquent les températures de nucléation et de croissance,(b) principe de la rampe de T(c) nombre de cristaux nucléés en function du temps (NaCl=0.7M
and pH=4.5 et (d) fréquence de nucléation en fonction de la sursaturation, à 20°C, à partir des pentes des droites de la figure c
( Article JCG 2010),
( Article CGD2011)
Cette dernière méthode a également des limites, elle sous-estime le nombre de cristaux nucléés. En effet, la taille critique varie avec la température, de sorte que lorsque la température change, la taille critique du nucleus change également : à la température de nucléation, la taille critique est plus petite qu'à la température de croissance. Par conséquent, si la température augmente jusqu'à la température de croissance, des germes plus gros que la taille critique à la température de nucléation, mais plus petits que celle de la température de croissance, seront dissous.
Expérimentalement notre choix s'est porté sur la seconde méthode. Dans un premier temps le montage de videomicroscopie multi-puits développé au laboratoire (fig.4) a été utilisé, il permet de réaliser jusqu'à 96 expériences en parallèle. Les résultats obtenus ont montré la difficulté de séparer la nucléation primaire homogène et hétérogène.
Approche microfluidique de la nucléation
Afin de limiter la nucléation aux interfaces nous nous sommes orientés vers l'utilisation des puces microfluidiques, qui permettent de générer des trains de gouttes (environ 200) de phase aqueuse dans l'huile dont le volume est de 250nL (fig.5-2). Les premiers résultats sont très prometteurs, en effet avec un outil simple d'utilisation et de conception nous avons pu mesurer des cinétiques de nucléation (fig.5-1c et d) avec de faibles quantités de matière première et nous avons également montré que les mécanismes qui gouvernent la nucléation à l'échelle du nL sont les mêmes que ceux dans des volumes plus élevés.

Figure 5-2 : Montage microfluidique en PDMS(a) puce avec canal de 500µm de diamètre, (b) module thermostaté,(c) et (d) gouttes et canaux,
(e)et (f) cristal de lysozyme dans une goutte.
( Article JCG2012)
Le montage microfluidique en PDMS a 2 limites clairement identifiées: (1) sa perméabilité qui devient importante au delà de 24h de stockage et (2) sa non compatibilité avec les solvants organiques. Nous avons donc mis en place un outil microfluidique simple et polyvalent a partir de tube en Teflon et de connections T en PEEK/Teflon qui permet de générer et stocker des gouttes, de 250 à 1nL, de phases aqueuses ou organiques dans une huile Teflon (figs. 5-3). Nous avons pu obtenir des cristaux dans des solvants organiques (fig.5-4)ou en milieu visqueux (fig.5-5)

Figure 5-3 : Montage microfluidique en PEEK/Teflon(a) Schema et photo de la junction T en PEEK, (b) formation de goutte d'éthanol dans huile fluorée avec junction T et (c) gouttes d'acétone dans une huile fluorée après 3 mois de stockage ( Article OPRD2012)

Figure 5-4 : Montage PEEK/Teflon (a) cristaux de lysozyme, (b) cristaux isonocotinamide dans éthanol et (c) cristaux de caféine dans éthanol dans une goutte ( Article OPRD2012)

Figure 5-5 : Montage PEEK/Teflon cristaux de protéine (Thèse S. Zhang) ( Article OPRD2015)
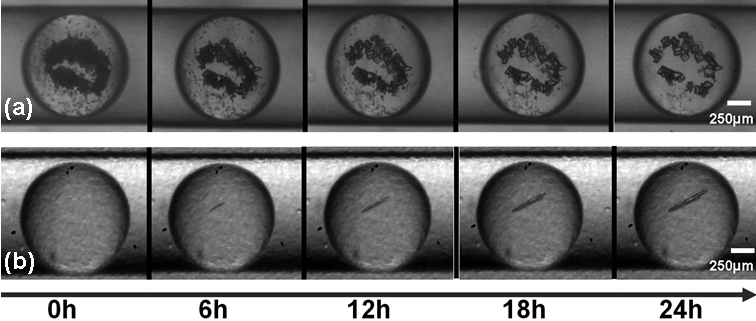
Figure 5-6 : Montage PEEK/Teflon cristaux de protéine (a) murissement d'Ostwald et (b) nucléation/croissance (Thèse S. Zhang) ( Article OPRD2015)
voir les videos Oswald et nucléationNous avons aussi montré l'aspect hétérogène de la nucléation et plus particulièrement le rôle de l'interface eau - huile CGD 2013
lien vers la thèse MANUEL ILDEFONSO
lien vers la thèse SHUHENG ZHANG
2.Approches Non-Classique de la Cristallisation
2.1. Approche non-classique: les champs externes
lien vers la these EVE REVALOR
Nous avons développé de nouvelles techniques de cristallisation sur de faibles volumes en présence d'un champ externe. Par champ externe, on entend en présence d'un faisceau lumineux (fig.6), d'un champ électrique (fig.7), magnétique, d'un point froid (respectivement chaud), ou d'ultrasons. Il s'agira d'être capable de provoquer la nucléation de manière localisée dans des solutions métastables. Deux effets sont attendus sur la structure des solutions sursaturées : orientation/structuration et fluctuations de densité. La théorie classique de la nucléation propose une nucléation en 1 étape : les fluctuations de densité et d'ordre étant simultanées. Un autre mécanisme proposé récemment suggère une nucléation en 2 étapes: d'abord une densification de la solution proche d'une démixtion puis une seconde étape où la phase dense s'ordonnerait.
- Irradiation lumineuse (article)
Le champ externe le plus documenté est l'irradiation lumineuse. Deux effets ont été reportés dans la littérature: d'abord, l'effet non-photochimique de la lumière sur la nucléation (NPLIN) à l'aide d'un laser et d'autre part, un effet photochimique induit par la lumière sur la nucléation (PLIN) à l'aide de lumière blanche (lampe au xénon). La NPLIN utilise un laser femto-seconde pour irradier la solution sursaturée, le mécanisme proposé est un alignement des molécules induit par le champ électrique du laser, l'effet Kerr optique. En ce qui concerne nos expériences, on utilise une lampe au Xenon pour irradier les solutions sursaturées de macromolécules biologiques, nous avons montré que non seulement l'irradiation provoque la nucléation dans la zone métastable, mais aussi qu'il existe un temps d'irradiation au-delà duquel la protéine est dénaturée. Nous avons alors réalisé un ensemencement de solutions non irradiées de lysozyme par de faibles quantités de solutions de lysozyme irradiées(figure 6), afin d'éviter la dénaturation des protéines par un excès d'irradiation. Nous avons montré qu'il s'agit d'un mécanisme d'activation photochimique de la nucléation . Dans le cas du lysozyme ce sont les radicaux formés au cours de l'irradiation qui accélèrent la nucléation par une augmentation sensible des interactions attractives intermoléculaires (montré par des mesures de diffusion quasi-élastique de la lumière) et formation de dimères comme précurseurs. Ces résultats ont ensuite été étendus à la thaumatine et au BPTI. Dans ce cas on peut parler de contrôle temporel, la nucléation est déclenchée grâce au champ appliqué à la solution métastable sursaturée.
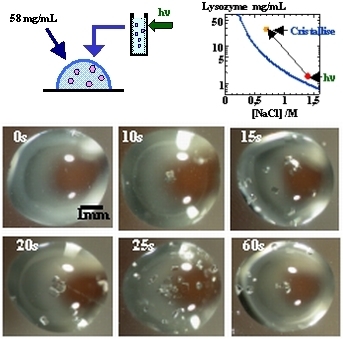
Figure 6 : Principe des expériences de cristallisation du lysozyme par irradiation lumineuse et photos de gouttes de cristallisation avec irradiation sous une lampe Xe 300V pendant 0, 10, 15, 20, 25 et 60s
- Champ electrique (article)
Nous avons observé une diminution du temps de nucléation, une augmentation de la cinétique de croissance et souvent un positionnement prédéterminé des cristaux. Dans la littérature la cristallisation des protéines contrôlée par un champ électrique est reliée à l'électromigration. Comme le BPTI, dans ces conditions, est chargé positivement à pH = 4,5 nous nous attendions à observer la cristallisation à la cathode. Nous avons observé une nucléation préférentielle des cristaux à l'anode, une croissance plus importante près de l'anode ainsi que la formation d'une phase autour de l'anode (fig.4), identifiée comme une phase riche en protéine due à une démixtion. Il existe, donc, un gradient de potentiel chimique du à l'intensité du champ électrique allant de l'anode vers la cathode, mis en évidence par: (1) la phase riche en protéine se trouve à l'anode, et (2) les cristaux sont plus gros près de l'anode, que de la cathode, ce qui est confirmé par la mesure des vitesses de croissance. On aurait pu penser que le gradient de concentration du au champ électrique serait plus intense au voisinage de la pointe (convergence des lignes de champs) induisant une localisation encore plus marquée. Cela n'est pas le cas en solution libre (fig.7), c'est dû à la convection qui s'oppose à l'effet du champ électrique en re-homogénéisant la solution. Afin de diminuer les mouvements de convection, nous avons réalisé l'expérience en gel d'agarose. La séquence de la figure 7 montre une excellente localisation de la nucléation au voisinage de la pointe pour le lysozyme et le BPTI.

Figure 7 : Montage et observations in-situ par microscopie optique de la cristallisation des protéines en présence d'une tension (Le + indique l'anode).
( CGD2007), ( CGD2009)voir la video
Toutes ces études trouvent aussi leur application dans la recherche de conditions de cristallisation de molécules bioactives ou d’intérêts pharmaceutiques, ainsi que pour les protéines solubles, virus et protéines membranaires, définition de protocoles rationnels pour la cristallisation, approche complémentaire aux méthodes statistiques proposées par les kits de cristallisation dite de cristallisation à haut débit ou high throughput screening (HTS).
2.2. Milieux Confinés
Ces dernières années nous avons assisté à un fort engouement pour la miniaturisation en cristallisation: outils microfluidiques, robots de criblage et cristallisation en émulsion. Ces méthodes de cristallisation en milieux confinés offrent des potentialités de contrôle intéressantes à la fois sur le plan des propriétés d'usage, mais également d'un point de vue de la sécurité et de l'environnement. Elles devraient permettre la conception de technologies innovantes pour la mise en forme et la stabilisation de cristaux permettant de fabriquer des matériaux à forte valeurs ajoutés. Ces techniques ont fait l'objet de nombreux travaux ces dernières années mais sont restées purement empiriques et n'ont pas conduit à la conception d'un procédé. Il manque donc des études locales quantitatives sur l'hydrodynamique (qui conditionne les transferts à l'échelle de la goutte), la thermodynamique (évolution des propriétés interfaciales au cours du procédé et diagrammes de phases…), et la cinétique (nucléation et croissance des cristaux en milieu confiné, stabilité des émulsions). Nous nous sommes intéressés à l'aspect thermodynamique du confinement dans un premier temps, le montage expérimental permettra dans un second temps d'aborder l'aspect cinétique.
(a) les milieux confinés: un modèle simple (article)
L'effet du confinement (solution de volume réduit) sur la nucléation/croissance a été abordé par le biais d'une formulation thermodynamique simple et concise. Nous pouvons ainsi spécifier la gamme de tailles de gouttes dans laquelle le confinement doit être pris en compte, ainsi que ses effets potentiels, parmi lesquels: unicité de l'événement de nucléation, accès à des sursaturations élevées impossibles en " bulk ", croissance explosive. Une expérience a été bâtie pour étudier le confinement et ses potentielles applications: contrôle de la nucléation, criblage, recherche de polymorphes,... En premier lieu, le confinement peut empêcher totalement la nucléation de se produire. Prenons l'exemple de la cristallisation dans une goutte. A sursaturations égales, deux gouttes de volumes différents ne contiennent pas le même nombre de molécules dissoutes. Comparons-le maintenant au nombre de molécules nécessaires à la création d'un seul cluster critique, de taille notée n*. Usuellement, le nombre de molécules en excès contenues dans une goutte sursaturée peut être considéré comme infini par rapport à n*; ce qui revient à l'hypothèse d'un réservoir infini assurant une sursaturation constante durant la nucléation. Mais qu'en serait-il pour une goutte, certes sursaturée, mais ne contenant qu'un nombre de molécules en excès égal à n* (le réservoir devient fini)? On conçoit aisément qu'il va falloir prendre en compte la variation de sursaturation au cours de la nucléation. Or, la taille de cluster critique n* est inversement proportionnelle à la sursaturation : plus la concentration dans la goutte diminue, plus le cluster critique à atteindre est grand et donc difficile à atteindre (fig.8a). Ainsi, pour une sursaturation initiale donnée, il existe une taille de goutte minimale, exprimée en Ns (Ns: représente le nombre de molécules que contiendrait cette goutte à saturation), de départ afin que la nucléation puisse prendre place. Les droites tracées sur la figure 8a, représente le chemin expérimental pour des tailles de cristallisoir de plus en plus petites (du noir au vert).Un second résultat important est obtenu : si la nucléation devient possible, alors il existe des tailles de gouttes où le cristal ne pourra croître au-delà d'une certaine taille, notée ici nstab*. La solution est ainsi toujours sursaturée, mais le cristal ne peut pas pour autant continuer sa croissance. Le cristal est en équilibre avec une solution sursaturée.
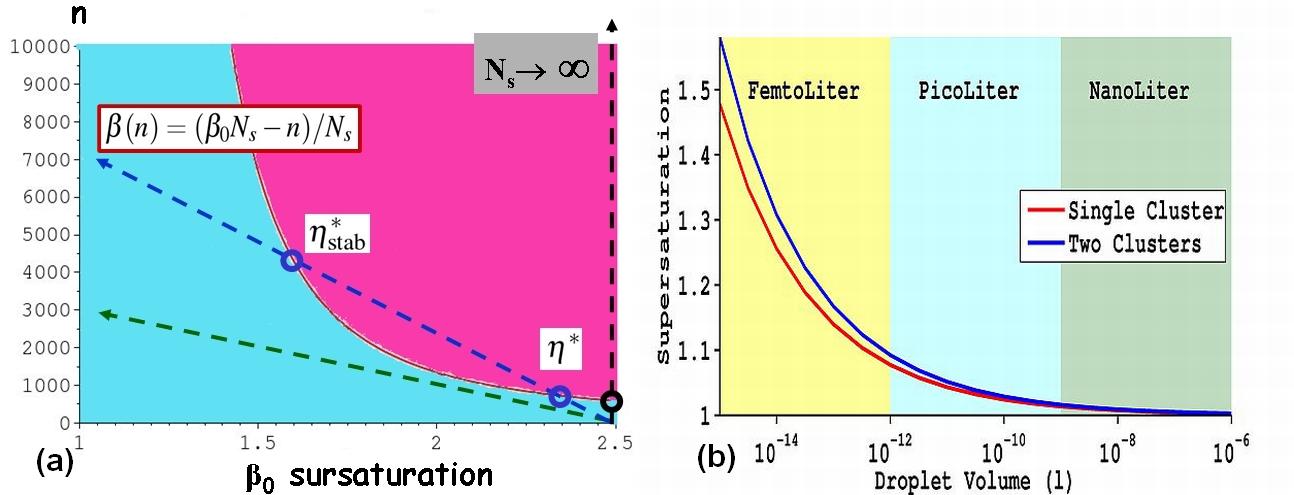
Figure 8 : (a)Variation du nombre de molécules constituant le germe critique en fonction de Bo pour une sursaturation initiale de 2,5, Ns représente le nombre de molécules que contiendrait cette goutte à saturation. Et B(n) la variation de sursaturation au cours de la nucléation. (b) Valeurs de la sursaturation initiale en fonction du volume de la goutte à partir de laquelle la nucléation devient possible (en rouge: 1 seul cristal, en bleu: 2 cristaux). Cas du lysozyme, pH=4,5, 0,7M NaCl à 20°C. (article)
On peut donc définir, maintenant, ce qu'est un milieu confiné, c'est le volume à partir duquel il est existe un effet du volume sur la nucléation qui dans le cas du lysozyme commence pour des volumes de l'ordre du pL (fig.8b). Le calcul a été effectué avec une hypothèse très simple sur la distribution des germes sous critique, il n'y'en a qu'un en regard des monomères. En prenant en compte la possibilité d'un deuxième germe, qui naitrait au même instant et croitrait à la même vitesse (cas peu probable, mais des plus restrictifs pour notre fenêtre d'unicité). On defini entre les courbes rouge et bleu de la figure 8b une fenêtre de nucléation d'un seul cristal. On peut donc espérer une fenêtre de validité de l'unicité de l'événement de nucléation plus étendue que celle calculée par cette méthode. Un autre résultat, au moins tout aussi important, est celui de la nature du cluster noté nstab* figure 8a : c'est le cluster critique stabilisé par l'effet de confinement. Pour résumer, le cristal stable en équilibre avec une solution encore sursaturée est en fait le cluster critique correspondant à cette sursaturation résiduelle, stabilisé par le biais du confinement, résultat que l'on retrouve en représentant l'énergie libre en fonction de n
(b) les milieux confinés: une expérience simple (article)
Les résultats, pour le moment issus de simulations, démontrent l'importance et l'originalité de cette recherche. Afin d'avérer expérimentalement de telles affirmations, une expérimentation a été développée. Il s'agit d'un générateur de microgouttes monté sur un microscope inversé (fig.9).
Figure 9 : (A)Montage expérimental pour générer des microgouttes: (a) microscope, (b) microcapillaire et (c) nanomanipulateur XYZ, (B)-(D) séquence montrant la création de gouttes, la taille augmente (de manière contrôlée) avec la vitesse de déplacement du microcapillaire. (article)
voir la video Generation de microgouttesLes gouttes de phase aqueuse sont générées sous huile par un microinjecteur utilisée en biologie (diamètre interne du capillaire 0,5µm), des nanodeplaceurs piézoélectriques assurent le positionnement et le déplacement du microcapillaire. En contrôlant la pression d'injection et la vitesse de déplacement on contrôle la taille des microgouttes qui sont générées dans le domaine du nL au pL (fig. 9B-D).
Nous avons pu générer des gouttes de NaCl de 3 à 1pL qui par diffusion de l'eau dans l'huile atteignent des sursaturations >90% (figures10a-c), alors qu'en milieux non-confinés la nucléation est spontanée pour des sursaturations de l'ordre de 3%! C'est l'effet cinétique du confinement. De plus une fois la sursaturation établie et après un temps de latence supérieure à 30 minutes, la croissance est ultra-rapide, de 0 à 15µm en moins de 2s. De nombreuses autres expériences sur KNO3, CaSO4.2H2O, la glycine et le BPTI ont confirmé ces résultats: les milieux confinés permettent d'atteindre des sursaturations élevées et on observe généralement un cristal par microgoutte. (JCG 2010)

Figure 10 : Séquence illustrant la nucléation du NaCl (a) à t=0 des microgouttes de 3 à 1pL
de solution 4,5M de NaCl sont générées, (b) t=4638s et (c) t=6120s.
(JCG 2010), (langmuir 2014)
voir la video évaporation de microgouttes
La séquence de la figure 10 illustre l'aspect aléatoire de la nucléation, on ne peut pas prédire quelle goutte va nucléer et à quelle moment. Le contrôle spatial de la nucléation est assuré par le milieu confiné (nucléation dans une goutte), mais il peut être amélioré et couplé au contrôle temporel si on arrive à contrôler le début de la nucléation. Pour cela on utilise les pointes réalisées au sein du département SSP (fig.7) pour déclencher la nucléation (figs. 10 b et c). La pointe est déplacée (en X,Y,Z) avec le même type de nanodéplaceurs que le microcapillaire qui a généré la goutte. (article) Le contact de la pointe avec la goutte va déclencher la nucléation et la croissance rapide (>200µm/s !!!) d’un seul cristal dans la goutte (fig.10d). Une approche tangentielle de la pointe par rapport à la goutte permet de fixer et manipuler le cristal (fig.10b)
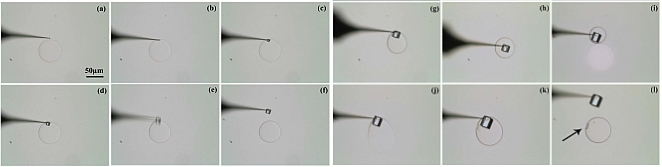
Figure 10_b : Séquence montrant la nucléation, la croissance et la manipulation d'un cristal de NaCl à sursaturation ?>1.97. La goutte contient à t=0 (a) une solution 0,7M NaCl et a une taille de 56 µm ( 51 pL) (a). Toutes les images sont au même grossissement. (article)
voir la video de la figure 10_b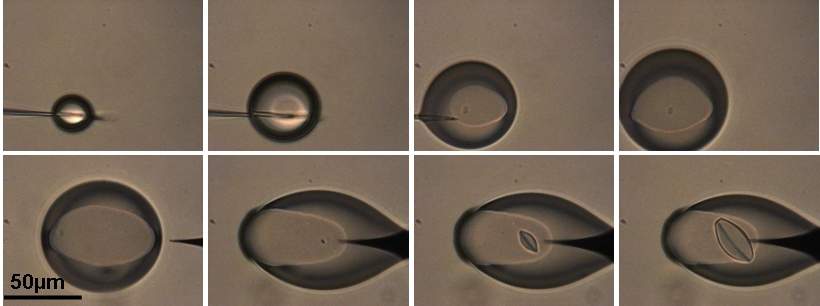
Figure 10_c : Séquence montrant la nucléation induite par une pointe et la croissance d'un cristal de protéine (BPTI). (article)

Figure 10_d : Séquence montrant la nucléation et la croissance rapide (>200µm/s entre images a et c) d’un cristal de NaCl, (a) condition initial: β>1.24, (a) taille de la goutte 60 µm (95pL). Toutes les images sont au même grossissement. Séquence réalisée avec une caméra sCMOS à 200images/s. (article)
3. Interactions Moléculaires, Structuration, Assemblage et Cristallisation des Biomolécules
(Article: Practical physics behind growing crystals of biological macromolecules)Les objectifs des travaux sur les biomolécules sont multiples: d'une part expliciter les mécanismes fondamentaux qui gouvernent l'auto-association, la nucléation et la croissance cristalline des systèmes biologiques complexes (protéines solubles, membranaires ou d'intérêt pharmaceutique), ce afin d'établir des protocoles rationnels de cristallisation pour l'obtention de monocristaux pour la cristallographie, mais également de maîtriser la cristallisation dans les procédés de production de principes actifs dans l'industrie pharmaceutique ; d'autre part de comprendre le rôle de l'environnement physico-chimique sur la structuration et l'association des complexes membranaires/tensio-actifs et des complexes biomolécules organiques/acides nucléiques. Le travail que nous réalisons consiste à déterminer des diagrammes de phases par mesure de solubilité, à mesurer de cinétiques de croissance par microscopie optique, à étudier les mécanismes de croissance cristalline par microscopie à force atomique, mais également en amont à étudier les interactions et l'assemblage de macromolécules en solution par diffusion de la lumière et diffusion des rayons X.
Température et Diagramme de Phases (voir c'est comprendre!)
(article sur effet de la température)
La visualisation des phénomènes est réalisée par le système monopuit thermostaté par effet Peltier couplé à un microscope inversé.
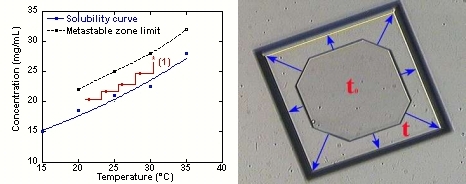
Figure 11: Croissance dans la zone metastable par contrôle de la température.
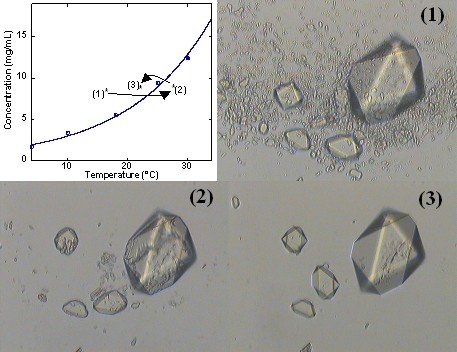
Figure 12: "Nettoyage" par murissement cinétique.

Figure 13: Contrôle du polymorphisme par variation de température. (article)
voir la video
Interactions, Diagramme de Phases et Nucleation (article)
On sait que la portée de l'interaction attractive nécessaire à la cristallisation influence le diagramme de phase. Une bonne connaissance des diagrammes de phases, du comportement en solution des macromolécules, du contrôle de la nucléation est donc nécessaire voire indispensable pour l'obtention de cristaux, car c'est le contrôle de ces mécanismes de croissance qui permettra la préparation de monocristaux de taille, de groupe d'espace adéquat et de qualité suffisante pour résoudre les structures cristallographiques par diffraction. On a ainsi pu caractériser avec différentes protéines, différentes zones dans le diagramme de phase : zone de cristallisation, zone de transition de phase liquide-liquide, phase gel. Les phénomènes de transition de phase peuvent influencer la cristallisation puisque l'on peut observer l'apparition de cristaux via la transition de phase liquide-liquide ou la phase gel. La thèse de S. Grouazel (2002-2005) a porté sur la caractérisation complète du diagramme de phases par des techniques de diffusion des rayonnements et par microscopie optique d'une petite protéine globulaire (fig. 13a), le BPTI, en présence de sel et sur l'influence des interactions, de l'auto-association des monomères de BPTI en décamères et d'une démixtion sur la cristallisation. Le couplage diffusion des rayons X aux petits angles et calculs de simulation numérique a permis de comprendre pourquoi la démixtion dans ce cas empêche la nucleation: les molécules de protéines ne sont pas agrégées mais sont en interactions répulsives et au contact (fig. 13b) et la forte viscosité du milieu diminue la diffusion des molécules comme dans l'état solide.
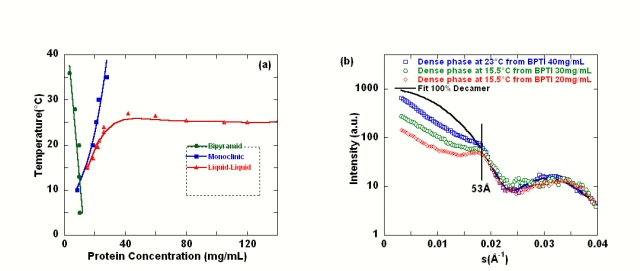
Figure 14: (a)Diagramme de phase du BPTI à pH 4,9 avec 350mM KSCN en fonction de la température et (b) sous la courbe de démixtion la phase dense est très concentrée et composée exclusivement de décaméres, le facteur de structure de la phase dense est de type répulsion de sphères dures, les molécules sont en contact (distance interparticulaire de 53 Å).
Afin de généraliser à un plus grand nombre de macromolécules biologiques les résultats fondamentaux de cristallisation obtenus par les différentes approches utilisées, nous avons dû utiliser les outils de la bio-informatique et créer sur le site du laboratoire une banque de données de cristallisation: la MPCD (http://www.cinam.univ-mrs.fr/mpcd/). Son analyse a permis de mettre en évidence des biais statistiques comme la température utilisée ou la nature des sels utilisés, des paramètres biochimiques non représentatifs, mais avant tout des paramètres pertinents pour la cristallisation comme le point isoélectrique ainsi que des tendances en accord avec les études faites depuis plusieurs années sur les interactions en solution et les paramètres qui contrôlent les potentiels attractifs de cristallisation.
Diffusion des RX aux petits angles (DXPA)
Depuis janvier 2008 nous sommes équipés d'un banc de DXPA, SWAXS system Hecus (fig.12), dispositif qui complète le banc de diffusion de la lumière Sem 633 (Sematech) déjà existant. La complémentarité des deux techniques est indispensable pour suivre le comportement des molécules en solution et réaliser l'étude des premiers stades de l'assemblage et/ou de la cristallisation des systèmes biologiques, afin d'en comprendre les mécanismes et de proposer des guides et des protocoles rationnels pour la cristallisation.
Figure 15 : Montage de diffusion des RX aux petits angles.
Les mesures de DXPA permettent d'accéder simultanément aux facteurs de forme et aux facteurs de structure des molécules en solution, sur un vaste domaine de vecteur de diffusion (angle), contrairement à la diffusion de la lumière qui ne permet de mesurer qu'un angle à la fois. Le facteur de forme donne accès à la taille (rayon de giration), à la masse et au degré d'association ou d'oligomérisation des molécules en solution (structure en solution) ; le facteur de structure à la distribution radiale des molécules en solution permettant par modélisation d'accéder aux potentiels de paires entre molécules.
CRISTALLISATION EN SOLUTION
Stéphane VEESLER (E-mail)
Directeur de recherche au CNRS (section 05)Recherche au sein du département Sources et Sondes Ponctuelles
contact : webmaster@cinam.univ-mrs.fr